
Le Pr Jun Ma (Université Sun Yat-sen) a présenté les résultats d'une étude de phase III, randomisée, multicentrique, en ouvert, qui a comparé la radiothérapie à la chimioradiothérapie concomitante dans le carcinome du nasopharynx (NPC) à risque moyen.
Radiothérapie vs chimioradiothérapie dans le carcinome du nasopharynx
La chimioradiothérapie concomitante (CCRT) avec ou sans induction ou chimiothérapie adjuvante est actuellement le traitement standard du NPC de stade II, sur la base d'une étude randomisée lors de laquelle la radiothérapie conventionnelle en deux dimensions était utilisée. Toutefois, à l'époque de la radiothérapie par modulation d'intensité (IMRT), nous ne disposions pas encore de preuves suffisantes concernant le rôle de la chimiothérapie dans cette population. En outre, une CCRT à base de cisplatine a accru les effets indésirables aigus graves, la toxicité tardive et le risque de décès lié au traitement. Une étude menée chez des patients atteints d'un NPC de stade II ou T3N0 a par ailleurs montré que l'ajout de la chimiothérapie concomitante à l'IMRT n'offrait pas d'avantage significatif en termes de survie (1).
Cette étude clinique de phase III, randomisée, multicentrique, doit démontrer si l'omission de la chimiothérapie concomitante est sûre pour les patients atteints d'un NPC à faible risque traité par IMRT. Le NPC à faible risque a été défini comme une maladie de stade II ou T3N0M0 sans les caractéristiques défavorables connues suivantes: diamètre maximal d'un ganglion lymphatique cervical > 3 cm, ganglions lymphatiques de niveau IV ou Vb, extension extraganglionnaire ou détection de > 4000 copies/ml d'ADN du virus d'Epstein Barr.
L'étude a randomisé 341 patients atteints d'un NPC à faible risque pour recevoir l'IMRT seule (n = 172) ou l'IMRT en association avec du cisplatine concomitant 100 mg/m2 par voie intraveineuse les jours 1, 22 et 43 (n = 169). Le critère d'évaluation principal de l'étude est la survie sans échec (failure-free survival, FFS), définie comme le temps jusqu'à la récidive (locorégionale ou à distance) ou jusqu'au décès. Les critères d'évaluation secondaires sont notamment la survie globale (OS), le profil de sécurité et la qualité de vie (HRQoL). Le suivi médian a atteint 46 mois. Dans le groupe CCRT, 60,4% des patients ont reçu trois cycles de chimiothérapie concomitante. 97,3% ont reçu au moins deux cycles.
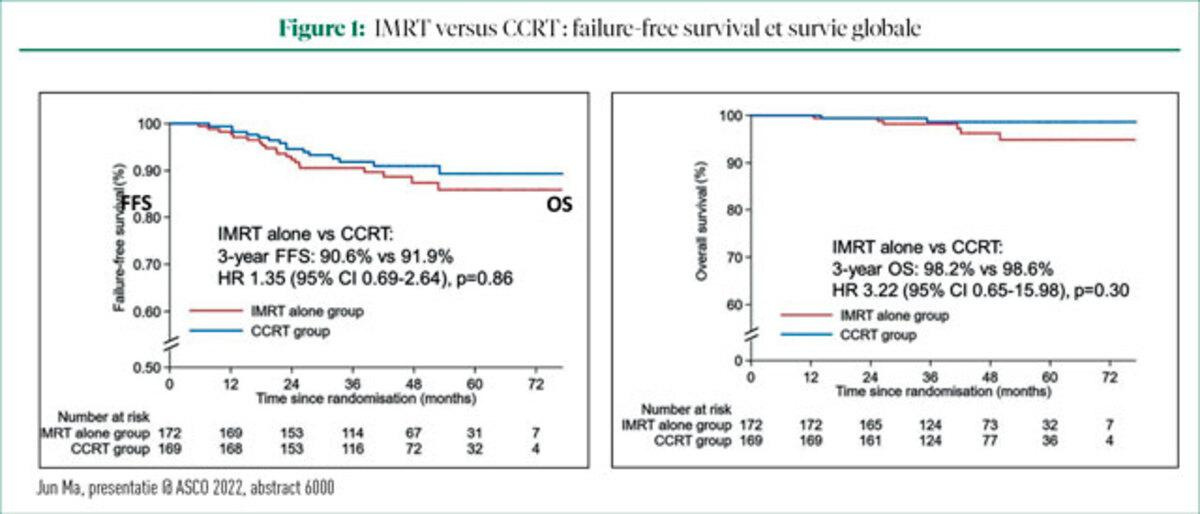
L'analyse du critère d'évaluation principal, la FFS, montre que le groupe ayant reçu uniquement l'IMRT présente une FFS à trois ans pratiquement identique au groupe CCRT. La FFS à trois ans s'élevait, respectivement, à 98,6% et 91,9%, ce qui correspondait à une marge de non-infériorité de moins de 10%. L'OS après trois ans était de 98,2% dans le groupe IMRT et de 97,6% dans le groupe CCRT (figure 1).
Pendant toute la durée de la cure de traitement, l'incidence des effets indésirables de grade III-IV rapportés était significativement plus faible dans le groupe ne recevant que l'IMRT (17%), par rapport au groupe CCRT (46,2%). La qualité de vie, mesurée à l'aide du questionnaire EORTC QLQ-C30, est significativement meilleure avec le traitement par IMRT seule, avec des différences nettes pour la fatigue, les nausées et vomissements, la douleur, l'insomnie, la diminution de l'appétit et la constipation.
Le Pr Jun Ma a dès lors conclu que l'IMRT seule est une option de traitement à part entière chez les patients atteints d'un carcinome du nasopharynx de stade II et T3N0. Ce traitement permet un contrôle de la maladie non inférieur, une diminution de la toxicité et une meilleure qualité de vie (2).
Le docétaxel comme radiosensibilisant
Le Dr Vanita Noronha (Tata Memorial Hospital, Mumbai, Inde) a présenté les résultats d'une étude randomisée de phase III sur l'utilisation du docétaxel comme radiosensibilisant chez les patients atteints d'un cancer de la tête et du cou qui ne sont pas éligibles à la chimioradiothérapie à base de cisplatine (CIS).
La chimioradiothérapie à base de CIS est le traitement standard pour les patients atteints d'un carcinome épidermoïde localement avancé de la région de la tête et du cou. Certains patients ne sont pas éligibles à un traitement par CIS. En outre, les données relatives à un traitement alternatif chez ces patients sont très limitées. Des études de phase I et II sur le docétaxel (DOX) se sont révélées prometteuses et la quasi-totalité des patients non éligibles au CIS peut recevoir du docétaxel. Des données d'études de phase III sont disponibles pour le cétuximab, le carboplatine et le 5FU, le carboplatine en monothérapie et l'association de paclitaxel et de carboplatine. Toutes ces données de phase III proviennent toutefois de cohortes de patients initialement éligibles au CIS. D'où la nécessité d'étudier le rôle du DOX chez des patients non éligibles au CIS.
Cette étude de phase III a inclus des patients adultes possédant un score de performance ECOG de 0 à 2, atteints d'un carcinome épidermoïde localement avancé de la région de la tête et du cou, qui étaient programmés pour une chimioradiothérapie en contexte adjuvant ou définitif. Les patients devaient être inéligibles au CIS. La non-éligibilité à un traitement par CIS était définie comme la présence d'au moins un des critères suivants: score de performance ECOG = 2 ; dysfonctionnement organique supérieur au grade II selon les CTCAE (Common Terminology Criteria for Adverse Events), version 4, par exemple une perte auditive, des acouphènes ou des troubles neurologiques ; hypersensibilité au CIS ; clairance de la créatinine calculée inférieure à 50 ml par minute, fonctionnement borderline d'un organe ou présence de comorbidités excluant l'utilisation du CIS ; perte de plus de 10% du poids corporel au cours des six mois précédents ; état de malnutrition défini comme un IMC < 16 kg/m2 ; et utilisation concomitante de médicaments néphrotoxiques nécessaires pour le traitement d'une affection concurrente.
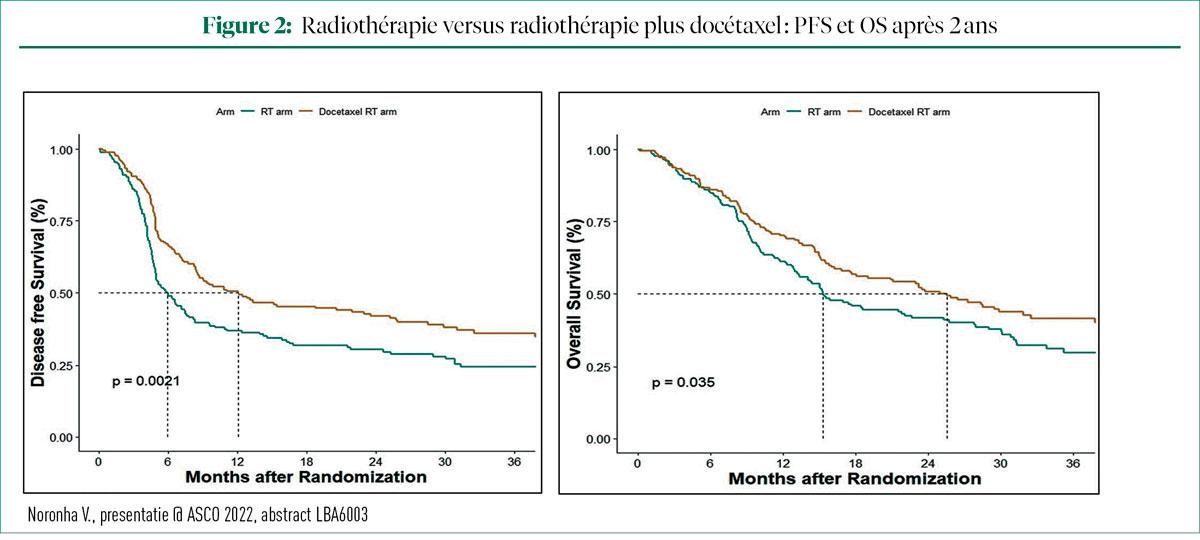
Les patients ont ensuite été randomisés selon un ratio de 1: 1 pour recevoir la radiothérapie (RT) seule ou l'association de DOX et RT. Le DOX était administré une fois par semaine à raison de 15 mg/m2, pendant toute la durée de la radiothérapie. Les critères d'évaluation de l'étude étaient la PFS après 2 ans, l'OS après 2 ans, la qualité de vie et les effets indésirables.
Le recrutement de cette étude s'est avéré lent, car elle a été menée au coeur de la pandémie de COVID. Au final, 777 patients ont été évalués afin de déterminer leur éligibilité et 356 ont été randomisés: 176 vers le bras RT seule et 180 vers le bras DOX + RT. La majorité des patients présentaient un stade IVA. Le statut HPV a été contrôlé chez environ la moitié des patients atteints d'une tumeur primitive de l'oropharynx. Le pourcentage de positivité au HPV était de 4%. 60% des patients ont reçu la chimioradiothérapie en contexte de traitement définitif et environ 40% l'ont reçue en contexte adjuvant. Un quart des patients n'était pas éligible à un traitement par CIS en raison d'une clairance de la créatinine faible. Environ 40% des patients présentaient une perte auditive de perception et environ 40% possédaient un score de performance ECOG de 2. Par ailleurs, la plupart des patients n'étaient pas éligibles au CIS pour différentes raisons. Plus de 90% des patients ont reçu 100% de la dose de RT, avec des interruptions de l'irradiation dans le bras DOX chez 11% des patients.
La médiane du nombre de cycles de DOX administrés était de six. Plus de 80% des patients ont reçu plus de cinq cycles. En ce qui concerne les effets indésirables, l'ajout de DOX à la RT a fait augmenter de manière significative le nombre de patients atteints de mucite, d'odynophagie et de dysphagie. Les autres toxicités, telles que la dermatite, les nausées, les vomissements et la neutropénie fébrile, n'étaient pas plus nombreuses à la suite de l'ajout de DOX.
Chez les patients ayant reçu la RT seule, la PFS après deux ans était de 30,3%. L'ajout de DOX a fait augmenter la PFS après deux ans à 42% (HR = 0,673 ; P = 0,002). L'amélioration de la PFS était cohérente dans tous les sous-groupes prédéfinis. L'OS après deux ans dans le bras RT était de 41,7%. L'ajout de DOX a fait augmenter l'OS après deux ans à 50,8%, soit un avantage absolu de 9% (HR = 0,747 ; P = 0,035). L'avantage en termes d'OS s'avère également cohérent dans tous les sous-groupes prédéfinis. Les scores relatifs à la qualité de vie étaient comparables dans les deux bras.
Cette étude présente toutefois quelques limites, abordées par le Dr Noronha. La majorité des patients de l'étude ont reçu une radiothérapie en deux dimensions. La CRT en trois dimensions ou l'IMRT réduit certes l'incidence de la xérostomie mais, chez les patients atteints d'un carcinome non nasopharyngé, il n'a pas été démontré que cela ait une grande influence sur l'efficacité. En outre, cette étude n'a pas fait de distinction entre les patients recevant une chimiothérapie en contexte adjuvant ou définitif. De plus, cette étude a été menée dans un seul centre, bien qu'il s'agisse d'un hôpital où le nombre de nouveaux patients atteints d'un cancer de la tête et du cou est supérieur à 10.000 par an.
Le Dr Noronha a conclu que l'ajout de docétaxel à la radiothérapie a amélioré la survie sans maladie et la survie globale chez des patients atteints d'un carcinome épidermoïde de la tête et du cou localement avancé non éligibles à un traitement par cisplatine. Elle estime que ce traitement peut désormais être considéré comme une nouvelle norme de référence de soins chez les patients non éligibles à la chimiothérapie à base de cisplatine (3).
Avasopasem manganèse: un nouveau traitement de la mucite buccale?
Le Dr Anderson (Université de l'Iowa) a présenté les résultats de l'étude ROMAN de phase III évaluant l'avasopasem manganèse (AVA) chez les patients qui développent une mucite buccale grave (SOM) sous chimioradiothérapie (CRT) pour un carcinome de la tête et du cou localement avancé non métastatique (LAHNC).
L'association IMRT + CIS est le traitement standard du LAHNC, durant lequel une majorité des patients développent une mucite buccale grave. Les critères de l'échelle de l'OMS relative à la mucite buccale reposent sur les ulcères visibles dans la cavité buccale et sur ce que le patient peut ingérer. La SOM est définie comme une mucite buccale de grade III ou IV. En présence d'un grade III, les ulcères buccaux contraignent le patient à ingérer uniquement des liquides. Dans le cas d'un grade IV, le patient est dépendant d'une sonde de nutrition. Environ 70% des patients atteints d'un LAHNC développent une SOM, dont 20% une SOM de grade IV. La durée médiane de la SOM est de 3 à 4 semaines, avec un début médian à partir de 40 Gy. Jusqu'à présent, il n'existe aucun médicament approuvé par la FDA dans la SOM.
L'avasopasem manganèse est un mimétique sélectif de la dismutase rapidement converti en peroxyde d'hydrogène sous l'action des superoxydes induits par le rayonnement. Ce point est important, car les superoxydes amorcent les dommages tissulaires et la cascade inflammatoire, entraînant ainsi le développement de la mucite buccale. Il a été montré lors de plusieurs études que l'AVA protège les cellules normales, mais pas les cellules cancéreuses, contre les effets nocifs du rayonnement. L'AVA a donné des résultats prometteurs lors d'une étude randomisée de phase II dans laquelle deux niveaux de dose, à savoir 30 mg et 90 mg, ont été testés contre placebo. La dose de 90 mg a réduit de manière statistiquement significative la durée de la SOM. Elle a également réduit l'incidence de la SOM induite par le rayonnement, ainsi que l'incidence de la mucite buccale de grade IV. Le profil des effets indésirables était comparable avec celui du placebo et, ce qui est plus important encore, l'efficacité du traitement était maintenue après un et deux ans (4).
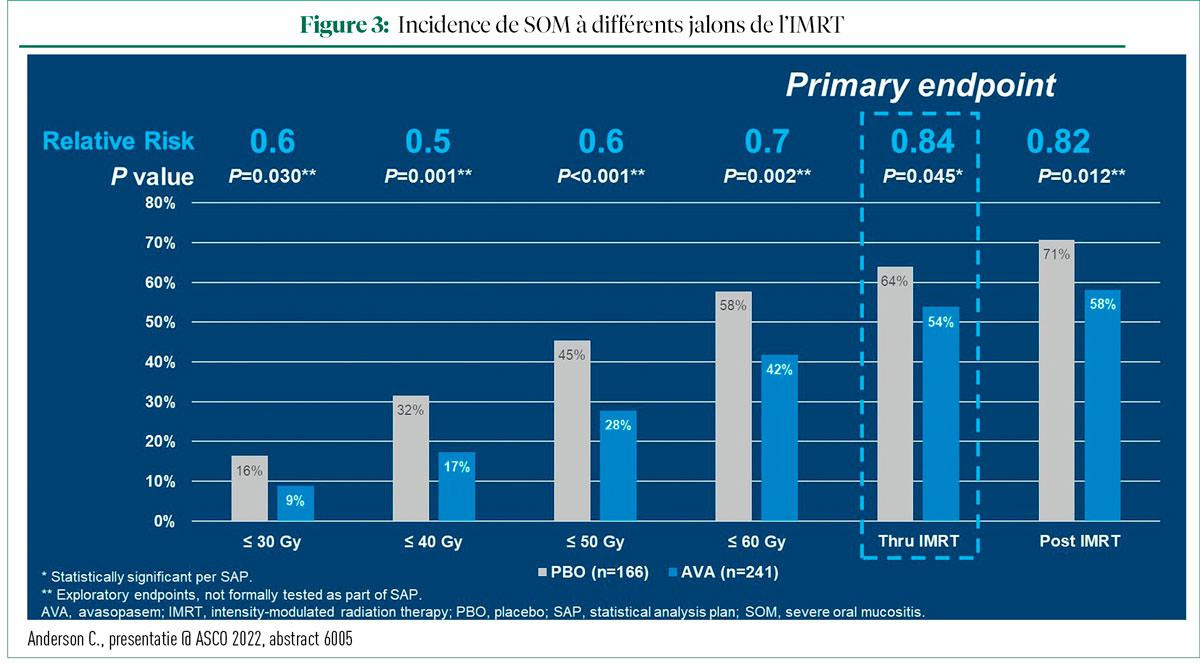
La dose de 90 mg a été choisie pour l'étude ROMAN de phase III, randomisée, en double aveugle. L'étude a inclus des patients atteints d'un carcinome épidermoïde localement avancé de la cavité buccale et de l'oropharynx, pour lequel une IMRT + CIS était prévue. Le traitement par AVA 90 mg ou placebo consistait en une perfusion intraveineuse de 60 minutes, préalablement à chaque fraction, du lundi au vendredi inclus, qui se terminait 60 minutes avant l'irradiation. Les patients étaient stratifiés sur la base de leur statut chirurgical et de leur schéma de cisplatine (toutes les trois semaines ou 40 mg/m2 toutes les semaines). Le critère d'évaluation principal de cette étude était l'incidence de la SOM en conséquence de l'irradiation. Les critères d'évaluation secondaires étaient le nombre total de jours de SOM ou la durée, ainsi que l'incidence et le nombre total de jours de mucite buccale de grade IV.
L'étude a randomisé 241 patients vers le bras AVA et 166 vers le bras placebo. Les caractéristiques des patients étaient bien équilibrées entre les deux bras, avec une majorité de patients positifs au HPV, atteints d'une tumeur primitive de l'oropharynx, qui ont reçu une chimioradiothérapie définitive selon un schéma hebdomadaire de CIS.
En ce qui concerne le critère d'évaluation principal des incidents de SOM à la suite de l'IMRT, il a été constaté que l'AVA réduisait de manière statistique l'incidence, de 64% à 54% (P = 0,045). En outre, l'AVA a réduit les incidents de SOM tant pendant la totalité de la phase d'irradiation que pendant la phase post-IMRT (figure 3).
Une impressionnante diminution de 56% de la durée médiane de la SOM a été observée, à savoir 18 jours dans le bras placebo contre seulement 8 jours dans le bras AVA. L'incidence de la mucite buccale de grade 4 a diminué, mais pas de manière statistiquement significative. En outre, l'AVA a retardé le début de la SOM de 38 jours dans le bras placebo à 49 jours dans le bras AVA. Chez les patients ayant reçu une cure complète d'AVA, c'est-à-dire 25 perfusions du placebo ou d'AVA, la diminution de l'incidence de la SOM était encore plus considérable: de 64% avec le placebo à 51% avec l'AVA.
L'AVA 90 mg s'avère bien toléré, ce qui est cohérent avec les données de l'étude de phase II. L'AVA provoquerait toutefois des nausées et vomissements légers, de grade 1 à 2, qui disparaissent spontanément.
Le Dr Anderson a conclu que l'AVA 90 mg est le premier médicament montrant une réduction statistiquement significative et cliniquement pertinente de l'incidence et de la durée de la mucite buccale grave avec, en outre, une amélioration significative de la gravité et une apparition retardée. Le profil de sécurité est comparable à celui du placebo (5).
1. Hunag et al., Front Oncol, 2020.
2. Jun Ma, presentation at ASCO's Annual Meeting 2022, abstract 6000.
3. Noronha V., presentation at ASCO's Annual Meeting 2022, abstract LBA 6003
4. Anderson et al., JCO 2019.
5. Anderson C., presentation at ASCO's Annual Meeting 2022, abstract 6005