La maladie de Huntington est une maladie génétique qui s'accompagne de troubles moteurs, cognitifs et/ou psychiatriques. Ses origines génétiques ont été identifiées en 1993. Comme chacun sait, il s'agit d'une maladie neurodégénérative monogénique. Des biomarqueurs obtenus en neuroimagerie montrent qu'une atrophie progressive du cerveau commence de nombreuses années avant l'émergence de la symptomatologie caractéristique de la maladie.
Joachim Ferreira (Lisbonne, Portugal)
25 ans après, où en est la recherche ?
Le développement de médicaments pour la maladie de Huntington, comme pour les autres maladies dégénératives, constitue un défi avec un niveau de déperdition élevé de molécules dans les phases précoces et un taux important d'échecs thérapeutiques dans les phases plus avancées des études cliniques.
Des essais déjà anciens ont été menés avec le riluzole contre un placebo par exemple. Les résultats n'ont montré aucune différence entre les deux bras de traitement. De même, comme l'a évoqué Joachim Ferreira (Lisbonne, Portugal) deux études ont été conduites, une aux Etats-Unis, une en Europe avec l'acide éthyl-eicosapentaénoïque sans résultat concluant. Un autre essai avec la latrepirdine s'est, lui aussi, révélé négatif, de même que celui de la pridopidine, de l'AFQO56 et de la cysteamine. Ces 8 études ont concerné un total de 1366 patients sur des périodes allant de 30 à 144 semaines.
Un espoir
Il existe actuellement deux médicaments qui ont montré une certaine efficacité pour réduire les symptômes hyperkinétiques : la tétrabenazine et la deutétrabenazine. Ces agents antagonisent de manière réversible la dopamine. Il s'agit donc bien de traitements symptomatiques. Ces deux médicaments sont autorisés par la FDA pour combattre la maladie de Huntington. La deutétrabenazine a été récemment testée contre placebo.
"Actuellement, nos données reposent sur des études contre placebo et pas en head-to-head", précise Joachim Ferreira. Les deux traitements sont assez comparables en ce qui concerne les effets sur la fonction motrice. Ils sont aussi comparables quant à leurs effets indésirables. La tétrabenazine et la deutétrabenazine n'ont pas des effets très différents en ce qui concerne les fonctions motrices et les effets indésirables. Il semble cependant exister une différence notable en ce qui concerne la dépression et la somnolence, beaucoup moins importantes avec la deutétrabenazine. Une étude clinique ouverte randomisée contrôlée évalue actuellement la possibilité d'une permutation de la tétrabenazine vers la deutétrabenazine.
Ongoing...
De nouveaux composés et stratégies thérapeutiques sont désormais en phase de développement clinique expérimentant de nouveaux mécanismes putatifs d'interventions. Certains ciblent des stratégies d'immunomodulation, mécanisme visant à réduire le niveau de la protéine mutante huntingtine (mHTT).
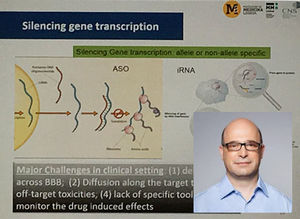
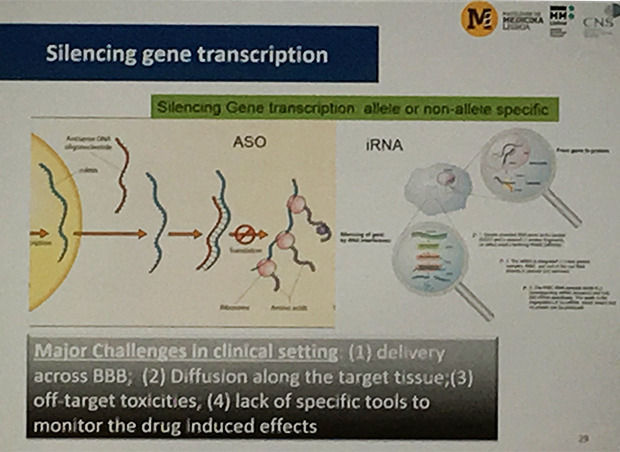
Une étude récente à l'échelle internationale a mis en avant 31 cas de maladie de Huntington testant 23 interventions différentes. Ces interventions sont encore à un stade d'essais cliniques. Ceux-ci explorent ce nouveau paradigme mêlant inhibition d'un agrégat de protéines mutantes huntingtine (PBT2), une sélection d'inhibiteurs SirT1 (selisistat), inhibition de PDE10A, des oligonucléotides antisense (ASO) interférant avec l'ARN, des anti-inflammatoires (laquinimod).
De nouvelles perspectives
Enfin, une nouvelle étude multicentrique et internationale se déroulant en Allemagne, au Canada, au Royaume-Uni est en cours et concerne un ASO : Ionis HTT. Cette première étude réalisée sur l'homme avec ce produit apporte une lueur d'espoir aux patients. En effet, les tests réalisés dans la première phase semblent confirmer jusqu'à présent stabilité et tolérance chez les patients. De plus, ces derniers ne présentent pas de complications face aux injections intrathécales.
C'est aussi la première fois qu'un médicament est utilisé pour l'inactivation de la transcription des gènes dans ce contexte. Il semble que la meilleure approche serait de limiter l'activation du gène codant pour la huntingtine à la fois dans le cortex et le striatum, tous deux touchés par la maladie de Huntington.
Il semble que le déclin lié à la maladie soit stoppé chez un grand nombre de patients de l'étude en cours. Si les essais de la première phase sont concluants, la deuxième phase devrait être lancée en 2018 incluant un plus grand nombre de patients.
La maladie de Huntington est une maladie génétique qui s'accompagne de troubles moteurs, cognitifs et/ou psychiatriques. Ses origines génétiques ont été identifiées en 1993. Comme chacun sait, il s'agit d'une maladie neurodégénérative monogénique. Des biomarqueurs obtenus en neuroimagerie montrent qu'une atrophie progressive du cerveau commence de nombreuses années avant l'émergence de la symptomatologie caractéristique de la maladie.25 ans après, où en est la recherche ?Le développement de médicaments pour la maladie de Huntington, comme pour les autres maladies dégénératives, constitue un défi avec un niveau de déperdition élevé de molécules dans les phases précoces et un taux important d'échecs thérapeutiques dans les phases plus avancées des études cliniques. Des essais déjà anciens ont été menés avec le riluzole contre un placebo par exemple. Les résultats n'ont montré aucune différence entre les deux bras de traitement. De même, comme l'a évoqué Joachim Ferreira (Lisbonne, Portugal) deux études ont été conduites, une aux Etats-Unis, une en Europe avec l'acide éthyl-eicosapentaénoïque sans résultat concluant. Un autre essai avec la latrepirdine s'est, lui aussi, révélé négatif, de même que celui de la pridopidine, de l'AFQO56 et de la cysteamine. Ces 8 études ont concerné un total de 1366 patients sur des périodes allant de 30 à 144 semaines. Un espoirIl existe actuellement deux médicaments qui ont montré une certaine efficacité pour réduire les symptômes hyperkinétiques : la tétrabenazine et la deutétrabenazine. Ces agents antagonisent de manière réversible la dopamine. Il s'agit donc bien de traitements symptomatiques. Ces deux médicaments sont autorisés par la FDA pour combattre la maladie de Huntington. La deutétrabenazine a été récemment testée contre placebo. "Actuellement, nos données reposent sur des études contre placebo et pas en head-to-head", précise Joachim Ferreira. Les deux traitements sont assez comparables en ce qui concerne les effets sur la fonction motrice. Ils sont aussi comparables quant à leurs effets indésirables. La tétrabenazine et la deutétrabenazine n'ont pas des effets très différents en ce qui concerne les fonctions motrices et les effets indésirables. Il semble cependant exister une différence notable en ce qui concerne la dépression et la somnolence, beaucoup moins importantes avec la deutétrabenazine. Une étude clinique ouverte randomisée contrôlée évalue actuellement la possibilité d'une permutation de la tétrabenazine vers la deutétrabenazine.Ongoing...De nouveaux composés et stratégies thérapeutiques sont désormais en phase de développement clinique expérimentant de nouveaux mécanismes putatifs d'interventions. Certains ciblent des stratégies d'immunomodulation, mécanisme visant à réduire le niveau de la protéine mutante huntingtine (mHTT).Une étude récente à l'échelle internationale a mis en avant 31 cas de maladie de Huntington testant 23 interventions différentes. Ces interventions sont encore à un stade d'essais cliniques. Ceux-ci explorent ce nouveau paradigme mêlant inhibition d'un agrégat de protéines mutantes huntingtine (PBT2), une sélection d'inhibiteurs SirT1 (selisistat), inhibition de PDE10A, des oligonucléotides antisense (ASO) interférant avec l'ARN, des anti-inflammatoires (laquinimod).De nouvelles perspectivesEnfin, une nouvelle étude multicentrique et internationale se déroulant en Allemagne, au Canada, au Royaume-Uni est en cours et concerne un ASO : Ionis HTT. Cette première étude réalisée sur l'homme avec ce produit apporte une lueur d'espoir aux patients. En effet, les tests réalisés dans la première phase semblent confirmer jusqu'à présent stabilité et tolérance chez les patients. De plus, ces derniers ne présentent pas de complications face aux injections intrathécales.C'est aussi la première fois qu'un médicament est utilisé pour l'inactivation de la transcription des gènes dans ce contexte. Il semble que la meilleure approche serait de limiter l'activation du gène codant pour la huntingtine à la fois dans le cortex et le striatum, tous deux touchés par la maladie de Huntington. Il semble que le déclin lié à la maladie soit stoppé chez un grand nombre de patients de l'étude en cours. Si les essais de la première phase sont concluants, la deuxième phase devrait être lancée en 2018 incluant un plus grand nombre de patients.J. Coutinho Ferreira - New therapeutic strategies in Huntington's disease - EAN 2017 Abstract #FW13_3